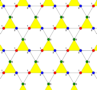
hi , I know the procedure for writing own unit cell i.e., you have to write lattice, latticegraph, unit cell, finite lattice, and graph. But I was trying for Kagome lattice but it wasn't working. So please help me out. I am giving you unit cell for kagome. Please find attached. Description: There are 5 vertices per unit cell. There is one triangle and a line included in that. The coordinates for 5 vertices are (0,0), (a,0) ,(a/2, sqrt(3)*a/2), (a/2, 0) and (a/4, sqrt(3)*a/4).
One more thing, how to extract the coordinates of any generated lattice? I am waiting for your reply.
Regards,
Aniket
Dear Aniket,
Why 5 sites in a unit cell? It seems to me that the three on a triangle are enough?
Matthias
On 26 Apr 2007, at 07:01, aniket@jncasr.ac.in wrote:
hi , I know the procedure for writing own unit cell i.e., you have to write lattice, latticegraph, unit cell, finite lattice, and graph. But I was trying for Kagome lattice but it wasn't working. So please help me out. I am giving you unit cell for kagome. Please find attached. Description: There are 5 vertices per unit cell. There is one triangle and a line included in that. The coordinates for 5 vertices are (0,0), (a,0) ,(a/2, sqrt(3)*a/2), (a/2, 0) and (a/4, sqrt(3)*a/4).
One more thing, how to extract the coordinates of any generated lattice? I am waiting for your reply.
Regards,
Aniket <kagome_1.png>
hi, I have attached unit cell of kagome. Please go through that and tell me how should I fill the information in lattice.xml file? Please tell me, how to extract the coordinates of any generated lattice?
Regards, Aniket
Dear Aniket,
Why 5 sites in a unit cell? It seems to me that the three on a triangle are enough?
Matthias
On 26 Apr 2007, at 07:01, aniket@jncasr.ac.in wrote:
hi , I know the procedure for writing own unit cell i.e., you have to write lattice, latticegraph, unit cell, finite lattice, and graph. But I was trying for Kagome lattice but it wasn't working. So please help me out. I am giving you unit cell for kagome. Please find attached. Description: There are 5 vertices per unit cell. There is one triangle and a line included in that. The coordinates for 5 vertices are (0,0), (a,0) ,(a/2, sqrt(3)*a/2), (a/2, 0) and (a/4, sqrt(3)*a/4).
One more thing, how to extract the coordinates of any generated lattice? I am waiting for your reply.
Regards,
Aniket <kagome_1.png>
If you look at your first picture you can see that the unit cell is a single triangle (yellow in your picture) and not the 5 sites shown in this message.
In the codes you can access the coordinates of the sites in the following way:
for (site_iterator it=sites().first ; it != sites().second ; ++it) { std::vector<double> c = coordinate(*it); }
Matthias
On 26 Apr 2007, at 08:56, aniket@jncasr.ac.in wrote:
hi, I have attached unit cell of kagome. Please go through that and tell me how should I fill the information in lattice.xml file? Please tell me, how to extract the coordinates of any generated lattice?
Regards, Aniket
Dear Aniket,
Why 5 sites in a unit cell? It seems to me that the three on a triangle are enough?
Matthias
On 26 Apr 2007, at 07:01, aniket@jncasr.ac.in wrote:
hi , I know the procedure for writing own unit cell i.e., you have to write lattice, latticegraph, unit cell, finite lattice, and graph. But I was trying for Kagome lattice but it wasn't working. So please help me out. I am giving you unit cell for kagome. Please find attached. Description: There are 5 vertices per unit cell. There is one triangle and a line included in that. The coordinates for 5 vertices are (0,0), (a,0) ,(a/2, sqrt(3)*a/2), (a/2, 0) and (a/4, sqrt(3)*a/4).
One more thing, how to extract the coordinates of any generated lattice? I am waiting for your reply.
Regards,
Aniket <kagome_1.png>
<kagome.pdf>
I am trying to run DMRG for the half-filled 4x4 Hubbard using Alps 1.3b and am getting energies which are substantially below the exact value. Granted, the jobs haven't finished, but all my (limited) knowledge of DMRG suggests that the energy will never drop below the exact value during any intermediate point in the calculation.
Hopefully someone can tell me what I am doing wrong with my input file or how my thinking is incorrect. I am confused that it got below E=-16 when the exact result is around E=-13.
Thanks,
Jeff
hubbard.parm: ========================================================= SWEEPS=10 MAXSTATES=1000 MODEL="fermion Hubbard" LATTICE_LIBRARY="/home/jeff/ALPS/lib/xml/lattices.xml" MODEL_LIBRARY="/home/jeff/ALPS/lib/xml/models.xml" CONSERVED_QUANTUMNUMBERS="Nup,Ndown" LATTICE="square lattice" {L=4;Nup_total=8; Ndown_total=8; t=1; U=4} =========================================================
This is how far I ran it before I killed it:
grep ITER *log
... ... ... WARMUP LEFT-TO-RIGHT ITERATION 12 ... ... ... WARMUP RIGHT-TO-LEFT ITERATION 12 ... ... ... WARMUP LEFT-TO-RIGHT ITERATION 12 ... RIGHT-TO-LEFT ITERATION 1 ITER = 1 ENTROPY = 1.00071007161 RIGHT-TO-LEFT ITERATION 2 ITER = 2 ENTROPY = 1.18324332788 RIGHT-TO-LEFT ITERATION 3 ITER = 3 ENTROPY = 1.24380103133 RIGHT-TO-LEFT ITERATION 4 ITER = 4 ENTROPY = 1.35051135715
And this is is where the energy was:
grep ener *log
... ... ... Iter = 2 Ener = -6.9064039895 Iter = 3 Ener = -9.65658596065 Iter = 4 Ener = -12.2462553335 Iter = 5 Ener = -13.9342133195 Iter = 6 Ener = -15.0550663096 Iter = 7 Ener = -15.6408884632 Iter = 8 Ener = -15.9408122129 Iter = 9 Ener = -16.1180047556
The exact result is -13.621854821 and Xiang's momentum space DMRG result is -13.571 (PRB 1996).
Hi Jeff,
I'll ask Adrian to look into this, but I wanted to warn you that DMRG does not work well for 2D lattices and also has problem with periodic boundary conditions. Are you trying to do 2D calculations with DMRG?
Matthias
On 26 Apr 2007, at 09:38, Jeff Hammond wrote:
I am trying to run DMRG for the half-filled 4x4 Hubbard using Alps 1.3b and am getting energies which are substantially below the exact value. Granted, the jobs haven't finished, but all my (limited) knowledge of DMRG suggests that the energy will never drop below the exact value during any intermediate point in the calculation.
Hopefully someone can tell me what I am doing wrong with my input file or how my thinking is incorrect. I am confused that it got below E=-16 when the exact result is around E=-13.
Thanks,
Jeff
hubbard.parm:
SWEEPS=10 MAXSTATES=1000 MODEL="fermion Hubbard" LATTICE_LIBRARY="/home/jeff/ALPS/lib/xml/lattices.xml" MODEL_LIBRARY="/home/jeff/ALPS/lib/xml/models.xml" CONSERVED_QUANTUMNUMBERS="Nup,Ndown" LATTICE="square lattice" {L=4;Nup_total=8; Ndown_total=8; t=1; U=4} =========================================================
This is how far I ran it before I killed it:
grep ITER *log
... ... ... WARMUP LEFT-TO-RIGHT ITERATION 12 ... ... ... WARMUP RIGHT-TO-LEFT ITERATION 12 ... ... ... WARMUP LEFT-TO-RIGHT ITERATION 12 ... RIGHT-TO-LEFT ITERATION 1 ITER = 1 ENTROPY = 1.00071007161 RIGHT-TO-LEFT ITERATION 2 ITER = 2 ENTROPY = 1.18324332788 RIGHT-TO-LEFT ITERATION 3 ITER = 3 ENTROPY = 1.24380103133 RIGHT-TO-LEFT ITERATION 4 ITER = 4 ENTROPY = 1.35051135715
And this is is where the energy was:
grep ener *log
... ... ... Iter = 2 Ener = -6.9064039895 Iter = 3 Ener = -9.65658596065 Iter = 4 Ener = -12.2462553335 Iter = 5 Ener = -13.9342133195 Iter = 6 Ener = -15.0550663096 Iter = 7 Ener = -15.6408884632 Iter = 8 Ener = -15.9408122129 Iter = 9 Ener = -16.1180047556
The exact result is -13.621854821 and Xiang's momentum space DMRG result is -13.571 (PRB 1996).
Matthias,
Yes, I'm trying to run the 4x4 square lattice with periodic boundary conditions (although I explicitly set the latter, perhaps that is an error with my input file).
I recognize that DMRG does not do well for 2D systems but I figured that it would just give me an upper bound, if just a very loose one. The naive DMRG for 2D lattices just converges very poorly in the number of states, m, and thus isn't very useful, but this does not explain how I get below the exact answer.
Thanks,
Jeff
Matthias Troyer wrote:
Hi Jeff,
I'll ask Adrian to look into this, but I wanted to warn you that DMRG does not work well for 2D lattices and also has problem with periodic boundary conditions. Are you trying to do 2D calculations with DMRG?
Matthias
On 26 Apr 2007, at 09:38, Jeff Hammond wrote:
I am trying to run DMRG for the half-filled 4x4 Hubbard using Alps 1.3b and am getting energies which are substantially below the exact value. Granted, the jobs haven't finished, but all my (limited) knowledge of DMRG suggests that the energy will never drop below the exact value during any intermediate point in the calculation.
Hopefully someone can tell me what I am doing wrong with my input file or how my thinking is incorrect. I am confused that it got below E=-16 when the exact result is around E=-13.
Thanks,
Jeff
hubbard.parm:
SWEEPS=10 MAXSTATES=1000 MODEL="fermion Hubbard" LATTICE_LIBRARY="/home/jeff/ALPS/lib/xml/lattices.xml" MODEL_LIBRARY="/home/jeff/ALPS/lib/xml/models.xml" CONSERVED_QUANTUMNUMBERS="Nup,Ndown" LATTICE="square lattice" {L=4;Nup_total=8; Ndown_total=8; t=1; U=4} =========================================================
OK, we'll look into it
Matthias
On 26 Apr 2007, at 10:09, Jeff Hammond wrote:
Matthias,
Yes, I'm trying to run the 4x4 square lattice with periodic boundary conditions (although I explicitly set the latter, perhaps that is an error with my input file).
I recognize that DMRG does not do well for 2D systems but I figured that it would just give me an upper bound, if just a very loose one. The naive DMRG for 2D lattices just converges very poorly in the number of states, m, and thus isn't very useful, but this does not explain how I get below the exact answer.
Thanks,
Jeff
Matthias Troyer wrote:
Hi Jeff, I'll ask Adrian to look into this, but I wanted to warn you that DMRG does not work well for 2D lattices and also has problem with periodic boundary conditions. Are you trying to do 2D calculations with DMRG? Matthias On 26 Apr 2007, at 09:38, Jeff Hammond wrote:
I am trying to run DMRG for the half-filled 4x4 Hubbard using Alps 1.3b and am getting energies which are substantially below the exact value. Granted, the jobs haven't finished, but all my (limited) knowledge of DMRG suggests that the energy will never drop below the exact value during any intermediate point in the calculation.
Hopefully someone can tell me what I am doing wrong with my input file or how my thinking is incorrect. I am confused that it got below E=-16 when the exact result is around E=-13.
Thanks,
Jeff
hubbard.parm:
SWEEPS=10 MAXSTATES=1000 MODEL="fermion Hubbard" LATTICE_LIBRARY="/home/jeff/ALPS/lib/xml/lattices.xml" MODEL_LIBRARY="/home/jeff/ALPS/lib/xml/models.xml" CONSERVED_QUANTUMNUMBERS="Nup,Ndown" LATTICE="square lattice" {L=4;Nup_total=8; Ndown_total=8; t=1; U=4} =========================================================
comp-phys-alps-users@lists.phys.ethz.ch
{kind=link}